
حدد اليوم الثامن من مايو من قبل منطمة الصحة العالمية ليكون اليوم العالمي للثلاسيميا ، ويهدف هذا اليوم إلى إرشاد المصابين بالثلاسيميا وتوعيتهم حول المرض ومضاعفاته وطرق علاجه ووقاية الأجيال القادمة منه ، رفع مستوى الوعي الصحي بمرض الثلاسيميا ، دعم المصابين بالثلاسيميا ومساعدتهم للتعايش مع المرض سواء صحيا أو نفسيا أو اجتماعيا إضافة إلى تشجيع المصابين بالمشاركة في توعية المرضى الآخرين وتبادل المعلومات الصحية ، السعي لحصول كل مريض على فرص الحصول على الرعاية الطبية الجيدة ، نشر المعلومات والتجارب والخبرات من البلدان التي لديها برامج مكافحة ناجحة للمحتاجين وتشجيع ودعم الدراسات والبحوث من أجل التحسين المستمر لاستراتيجيات الوقاية والرعاية الطبية والعلاج .
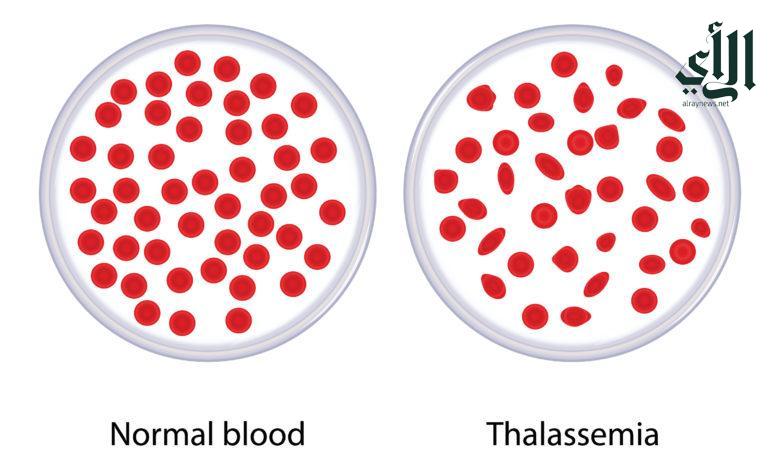
تعد الثلاسيميا من الاضطرابات الوراثية في خلايا الدم ، ويوصف بإنخفاض عدد كريات الدم الحمراء عن المعدل الطبيعي وأيضاً إنخفاض مستوى الهيموجلوبين ، كما أن هناك مسميات أخرى منها ( أنيميا البحر الأبيض المتوسط ) ، وذلك لكثرة انتشاره في منطقة حوض البحر الأبيض المتوسط .
أنواع الثلاسيميا
تتعدد أنواع الثلاسيميا اعتماداً على عدد الطفرة الجينية ، وعلى الجزء المصاب بها ، حيث أن الطفرة تحدث في أجزاء الهيموجلوبين ألفا أو بيتا أو كلاهما :
ثلاسيميا ألفا حيث يتكون الهيموجلوبين من أربع سلاسل جينية من النوع ألفا ( اثنتين من الأب و اثنتين من الأم ) ، وعند حدوث خلل أو قصور في هذا السلاسل ينتج ما يسمى ( ثلاسيميا ألفا ) ، وتختلف حدتها حسب درجة الخلل أو القصور ، فعند حدوث خلل في سلسلة جينية واحدة تسمى ( الثلاسيما الساكنة ) ، ويعد الشخص حاملاً للجين المصاب ولا يعاني المصاب أعراضاً ظاهرة ، أما عند خدوث خلل في سلسلتين او جينيتين من النوع ألفا تنتج حالة ( الثلاسيميا ألفا البسيطة ) ، ويعاني الشخص الحامل لهذه الجينات أعراضاً بسيطة جداً ، وقد لا تكون ظاهرة ، لكن يمكن اكتشافها من خلال فحص الدم ، وفي حالة حدوث القصور في ثلاث سلاسل جينية من ألفا ينتج فقر دم شديد ، وتتراوح الأعراض التي يعانيها الشخص ما بين المتوسطة إلى الشديدة وتسمى الحالة ( مرض الهيموجلوبين هـ ) ، ويظهر تحليل الدم للمصاب في هذه الحالة كريات دم حمراء صغيرة و مشوهة ، كما يصاب المريض أيضاً بتضخم في الطحال وتشوه في العظام ؛ بسبب زيادة نشاطها لتعويض الخلايا الحمراء التالفة ، مما يجعله ذلك في حاجة إلى نقل الدم ليتمكن من الحياة بشكل طبيعي ، وعند حدوث الخلل أو القصور في أربع سلاسل جينية فتسمى (الثلاسيميا ألفا الشديدة) ، وتتسبب في وفاة الحنين قبل الولادة أو مباشرة بعد الولادة .
ثلاسيميا بيتا و هنا يتكون الهيموجلوبين من سلسلتين من النوع بيتا ، تورث كل سلسلة من أحد الأبوين ، وتنقسم حالات الإصابة إلى قسمين وذلك حسب عدد السلاسل التي يحدث فيها الاضطراب :
الثلاسيميا الصغرى و تحدث بسبب حصول اعتلال في إحدى السلاسل الجينية فقط و لا يعاني المصاب أعراض ظاهرة سوى فقر دم بسيط يظهر أثناء التحاليل الروتينية للدم .
الثلاسيميا الكبرى تحدث عند حدوث الخلل في السلسلتين الجينيتين ، و يعاني المصاب أعراض فقر دام شديدة و تشوهاً في العظام و تضخماً في الطحال ، ولذلك فهو بحاجة لنقل الدم بشكل منتظم ليتمكن من الحياة بشكل طبيعي ، ولا تظهر هذه الأعراض عند ولادة الطفل ، ولكنها تبدأ في الظهور خلال العاميين الأوليين من العمر.
السبب و عوامل الخطورة:
تحدث الثلاسيميا بسبب الطفرة الجينية في الحمض النووي للخلايا المكونة للهيموجلوبين ، و تنتقل هذه الطفرة وراثياً من الأباء إلى الأبناء ، مما يتسبب حدوث الطفرات الجينية في تعطيل إنتاج الهيموجلوبين الطبيعي ، وبالتالي فأن انخفاض مستويات الهيموجلوبين وارتفاع معدل تلف خلايا الدم الحمراء ( وهو ما يحدث لدى مرضى الثلاسيميا ) ، يؤدي إلى ظهور أعراض فقر الدم .
وتكمن عوامل الخطورة في وجود تاريخ عائلي للإصابة بالثلاسيميا ، كما يؤثر الثلاسيميا بشكل أساسي في الأشخاص الذين لديهم أفراد من العائلة في الأصل من حول البحر المتوسط بما في ذلك ، إيطاليا ، اليونان ، قبرص ، الهند ، بنغلاديش ، والشرق الأوسط والصين وجنوب شرق آسيا.
التشخيص و أهمية الفحص ما قبل الزواج:
تذكر لنا د. شيماء آل عون ، أخصائية أمراض وأورام الدم بمدينة الملك فهد الطبية بالرياض ، أن الثلاسيميا تعد من أشهر أمراض الدم الوراثية بعد مرض الأنيميا المنجلية ، يصنف انتشارها حول العالم ٥٪ ، وللأسف لا يوجد أحصائيات حديثة ف المملكة العربية السعودية لكن تسجل أعلى المعدلات في المنطقة الشرقية والجنوبية.
وفي الجانب التشخيصي ذكرت أن التشخيص يتم عن طريق استخدام اختبارات الدم ، بما في ذلك اختبار صورة الدم الكاملة ، قياس نوع الهيموجلوبين، اختبار كمية الحديد في الدم لمعرفة ما إذا كان فقر الدم بسبب نقص كمية الحديد أو بسبب الثلاسيميا ، الدراسات الوراثية لتحديد نوع وشدة الإصابة.
وأضافت أن مرض الثلاسيميا هو أحد اضطرابات الدم الوراثية ، ينتقل من الآباء للأبناء ، وتعتمد شدة الأعراض على عدد وموقع الطفرات الجينية التي ورثها الطفل من الأهل . وكلما زادت الجينات الطافرة زادت شدة الثلاسيميا ،لذلك اكتشاف الإصابة في سن متأخر قد يكون بسبب عدم ظهور الأعراض في سن مبكرة بسبب نوعية الطفرة كون المصاب حاملاً للمرض أو مصاب ثانوي .
وعن أهمية الفحص ما قبل الزواج ذكرت يجب إجراء فحص الثلاسيميا قبل الزواج للتأكد من عدم إصابة الزوجين بالمرض ، أو حمل الجين المصاب عند كليهما ؛لأنه وفي حال كان الزوجان حاملان لجين المرض بالرغم من عدم الإصابة فإن احتمال إنجاب طفل مصاب بالثلاسيميا يعادل نسبة 25% ، لذلك يجب توعية الزوجين حول المرض وطبيعته وصعوبته هو شرط أساسي لإتمام الزواج ، والحد من إنجاب المزيد من الأطفال المصابين بالثلاسيميا.
و أختتمت بـ يجب على مرضى الثلاسيميا الإلتزام بالمتابعة مدى الحياة مع طبيب الدم ، فالمرض مزمن يتطلب الفحوصات ونقل الدم بشكل دوري ، وأخذ الأدوية المقللة لكمية الحديد في الجسم لما قد يترتب عليه من فشل باقي الأعضاء بسبب هذا التراكم نتيجة نقل الدم المتكرر ، كذلك أخذ التطعيمات و اللقاحات للوقاية من العدوى و الالتهابات ، ومراجعة الطبيب قبل أي اجراء جراحي أو أخذ مكملات غذائية ، و بالنسبة للمرأة الحامل يجب مراجعة طبيب أمراض الدم طوال فترة الحمل حتى وإن كانت حاملة فقط للمرض ، وأخيراً ، يجب توعية بقية أفراد المجتمع بالمبادرة بالتبرع بالدم لما فيه من أجر وإنقاذ حياة لهؤلاء المصابين وغيرهم فهو أقل ما يمكن عمله لمساندتهم .
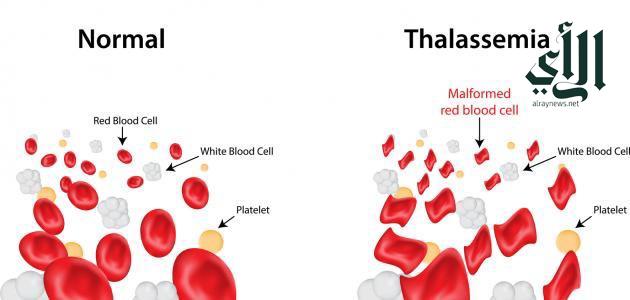
الأعراض و المضاعفات .. الوقاية و العلاج:
ذكرت د. منى عسيري ، استشاري أمراض دم و أورام أطفال ، رئيس وحدة أمراض الدم و أورام الأطفال بمستشفى أبها للولادة و الأطفال ، أن نسب الإصابة باضطراب الثلاسيميا عالميا تتراوح ما بين الـ 100 ألف طفل مصاب إلى 200 ألف سنوياً ، و تزيد النسبة في دول البحر المتوسط و دول الشرق الأوسط ، والجنوب الغربي من قارة آسيا ، و في المملكة العربية السعودية تشكل نسبة المصابين بـ الثلاسيميا تصل إلى 13٪ سنويا من عدد أمراض الدم في المملكة ، و تتركز في المنطقة الشرقية و الشمالية و الجنوبية خصوصاً منطقتي محايل عسير و جازان .
و أضافت أن هناك عدد كبير من مرضى ( الألفا الثلاسيميا ) و تعتبر الأقل انتشاراً عالمياً من البيتا أو أنيميا البحر المتوسط ب ، كما أنه قد تم تسجيل عدد من الإصابات لدى عدد من العوائل في جنوب المملكة .
كما قالت بأن التحليل ما قبل الزواج هو السبيل الوحيد في الوقاية من تناقل المرض ، و ذكرت بأن الأعراض غالباً تبدأ بالظهور في عمر 6 شهور ، عندما يختفي الهيموجلوبين الجنيني ، و من الأعراض التي قد تظهر الإرهاق ، الضِّعف ،شحوب الجلد و اصفراره ، انتفاخاً في البطن نتيجة تضخم الكبد و الطحال و البول الداكن ، و مع تقدم العمر يحدث تشوهات لعظام الوجه و بطىء النمو ، و من المضاعفات التي قد تصيب المريض ، مضاعافات المرض نفسه بطىء النمو ، تشوهات في عظام الوجه ، الاعياء والتعب ، و مضاعفات نقل الدم المتكررارتفاع نسبه الحديد وترسبها على اعضاء مهمه في الجسم على البنكرياس وغيرها .
و من الجانب العلاجي ذكرت بأن العلاج المتاح هو نقل الدم بشكل دوري بمعدل كل ٣ الى ٤اسابيع ، بالإضافة إلى متابعة نسب الحديد وفي حال ارتفاعه البدء بـ أدوية منع ترسب الحديد ، و المتابعة المستمرة مع طبيب الدم لمتابعة النمو و وظائف الكبد والكلى بشكل دوري ، كما ذكرت أيضا أن العلاج النهائي هو زراعه نخاع العظم ، وهي عملية تقدم في المستشفيات المتقدمة في المملكة العربية السعودية و نسب النجاح ممتازة مقارنة بنسبتها عالميا وهذا بفضل الله ثم بفضل كفاءة الأطباء والكوادر الطبية في تلك المراكز .
و أنهت حديثها بـ نصيحتي لحاملي مرض أنيميا البحر المتوسط أو أي مرض يتم اكتشافه في تحليل ما قبل الزواج بعدم إكمال الزواج ، حيث أن خطوره الإصابة بالمرض قد تصل لـ 25٪ في كل حمل ، وبـ إنجاب طفل مصاب ف أن هذا يكبد الأسرة الكثير من التعب النفسي والجسدي والمادي وغيره ، ويكبد المستشفيات زياده في اعداد المرضى ، و أضافت أما المرضى المصابين أنصحهم بالإستمرار في المتابعة المستمرة والأخذ بنصائح الأطباء والحمدلله العلاج متاح للجميع .
 (
( (
(